Programas
Molecular Dynamics
Molecular Dynamics
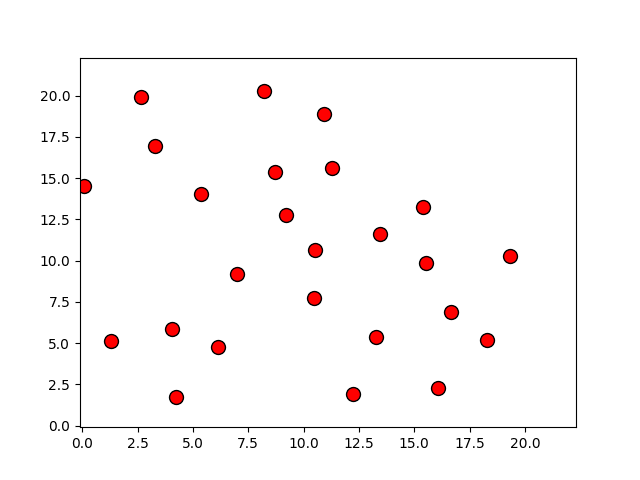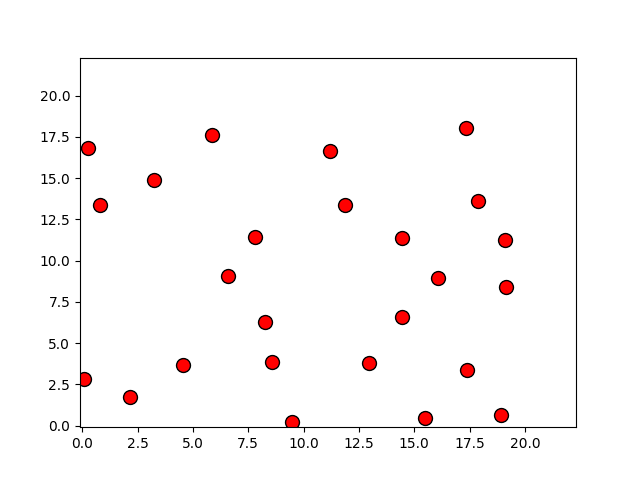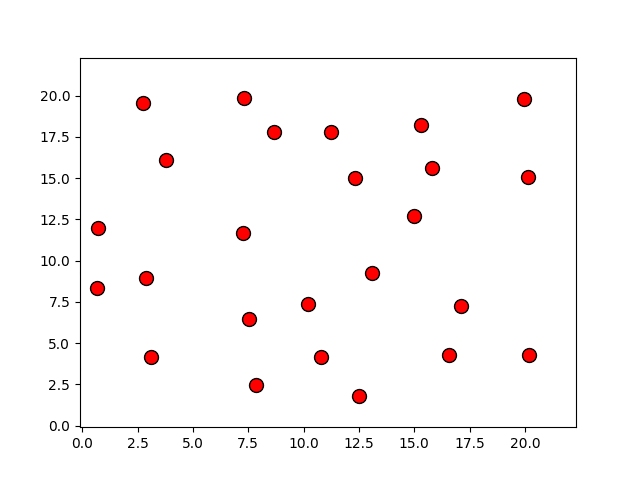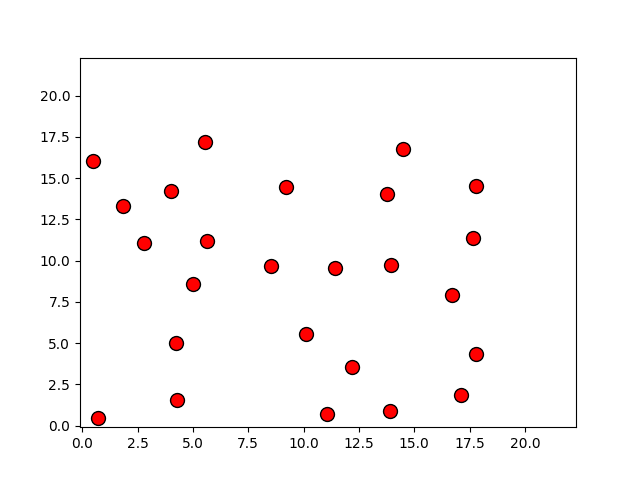 |
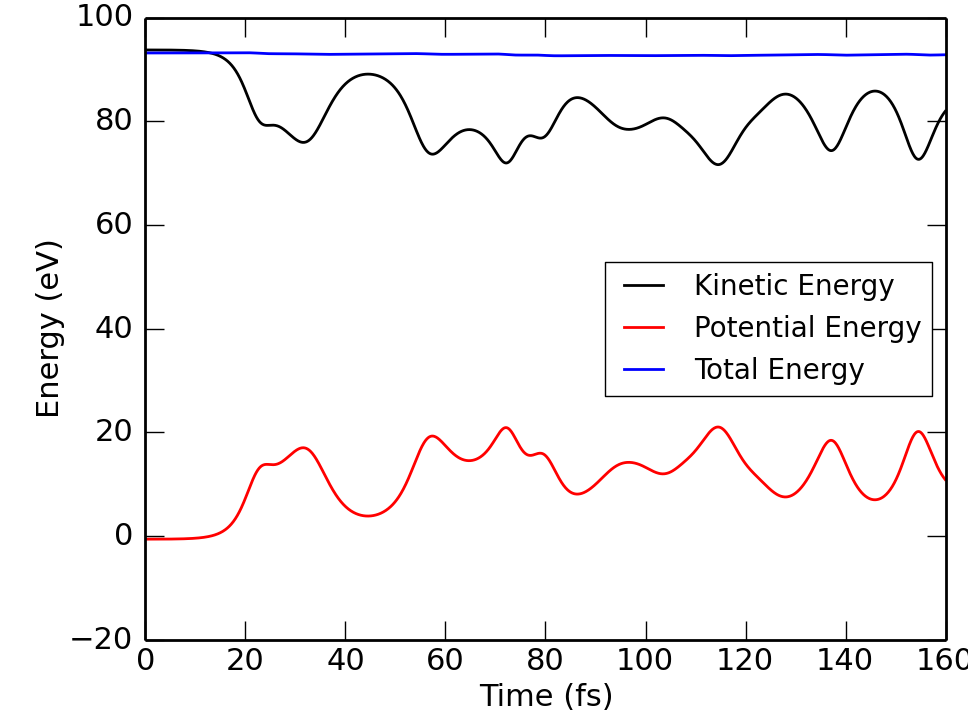 |
INDEX
Code (python)
#!/usr/local/bin/python
#***********************************************************************#
#* MOLECULAR DYNAMICS *#
#* *#
#* This program performs molecular dynamics for an argon 2D-crystal *#
#* *#
#* For B. Militzer course, EPS109, UC Berkeley, 2018. *#
#***********************************************************************#
# AUTHOR: FELIPE GONZALEZ CATALDO, September 2018.
from pylab import *
# Physical parameters
epsilon = 0.0103 # LJ potential Argon
sigma = 3.41 # LJ potential Argon
m = 39.948 # Argon Mass
dt=0.2 # in fs ( 1 fs = 10^-15 s)
# Lattice
nx=5
ny=5
a0 = 4.056
L = [ nx*a0, ny*a0 ]
# MD
nBlocks = 400
PBC = True # Periodic boundary conditions
r = array([ [a0*i,a0*j] for i in range(nx) for j in range(ny) ])
v = array([ [random(), random()] for i in range(nx) for j in range(ny) ]); v= (2*v-1)/20.0
a = array([ [0.0,0.0] for i in range(nx) for j in range(ny) ])
a_old = array([ [0.0,0.0] for i in range(nx) for j in range(ny) ])
'''
EXAMPLE 1:
'''
#nx=2; ny=1; dt= 2.0; a0 = 1.2*sigma; PBC= False; nBlocks= 400
#r = array([ [2, 2], [2+a0, 2] ])
#v = array([ [0.002,0.0], [-0.002,0.0] ] )
'''
EXAMPLE 2:
'''
dt = 1.0; nBlocks=400
v = array([ [random(), random()] for i in range(nx) for j in range(ny) ]); v= (2*v-1)/20.0
'''
EXAMPLE 3:
'''
#dt = 0.2
#v = array([ [random(), random()] for i in range(nx) for j in range(ny) ]); v= (2*v-1)/20.0
NATOMS= len(r)
def LennardJones(r): # r is the collection of all positions ri=(xi,yi)
U = 0.0
for i in range(NATOMS):
for j in range(i+1,NATOMS):
dr = r[i] -r[j]
if PBC:
if ( dr[0] >= 0.5*L[0] ): dr[0] -= L[0]
elif ( dr[0] < -0.5*L[0]): dr[0] += L[0]
if ( dr[1] >= 0.5*L[1] ): dr[1] -= L[1]
elif ( dr[1] < -0.5*L[1]): dr[1] += L[1]
rij = norm(dr)
U += 4.0*epsilon*( (sigma/rij)**12 - (sigma/rij)**6 )
return U
def KineticEnergy(v):
#v2 = [ dot(vi,vi) for vi in v ]
#sum_v2 = reduce((lambda x, y: x + y), v2)
#return 0.5*m*sum_v2*103.6427 # amu * (angstrom/fs)^2 = 103.6427 eV
v2 = 0.0
for vi in v:
v2 += norm(vi)**2
return 0.5*m*v2*103.6427
def Force(ri,rj):
dr = ri -rj
if PBC:
if ( dr[0] >= 0.5*L[0] ): dr[0] -= L[0]
elif ( dr[0] < -0.5*L[0]): dr[0] += L[0]
if ( dr[1] >= 0.5*L[1] ): dr[1] -= L[1]
elif ( dr[1] < -0.5*L[1]): dr[1] += L[1]
rij = norm(dr)
coeff = (sigma/rij)
force = -4*epsilon*( 12*coeff**11 - 6*coeff**5 ) * (-coeff/rij)*dr/norm(dr)
return force # in eV/angstrom
fig = figure(1)
ax = subplot(111)
xlim(-0.1, 1.1*L[0])
ylim(-0.1, 1.1*L[1])
#xlim(min(r[:,0]), max(r[:,0]),)
#ylim(2*(min(r[:,1]) -1), 2*(max(r[:,1])+1) )
p, = plot(r[:,0], r[:,1], 'o')
show(block=False)
"""
MOLECULAR DYNAMICS
"""
for iBlock in xrange(nBlocks):
''' PRINT PROPERTIES '''
print "t[fs]=",iBlock*dt, "Kin[eV]=", KineticEnergy(v), "Pot[eV]=", LennardJones(r)
for i in range(NATOMS):
a[i] = 0*a[i]
# Force over atom i
for j in range(NATOMS):
if i != j:
a[i] += 0.0096485333*Force(r[i],r[j])/m # <---- Inefficient, repited division and units conversion ( Force/mass = (eV/A)/(amu) = 0.0096485333 A/fs^2 )
'''
Euler-Cromer algorithm
'''
#####
v[i] = v[i] + dt*a[i]
r[i] = r[i] + dt*v[i]
####
'''
Velocity Verlet algorithm
'''
#####
#r[i] = r[i] + dt*v[i] + 0.5*dt*dt*a[i]
#v[i] = v[i] + dt*0.5*(a[i] + a_old[i])
#a_old[i] = a[i]
####
if PBC:
for q in range(2):
if r[i][q] >= L[q]: r[i][q] -= L[q]
elif r[i][q] <= 0.0: r[i][q] += L[q]
p.set_xdata(r[:,0])
p.set_ydata(r[:,1])
plt.pause(1e-30)
draw()
#savefig(str(iBlock)+'.png')
#***********************************************************************#
#* MOLECULAR DYNAMICS *#
#* *#
#* This program performs molecular dynamics for an argon 2D-crystal *#
#* *#
#* For B. Militzer course, EPS109, UC Berkeley, 2018. *#
#***********************************************************************#
# AUTHOR: FELIPE GONZALEZ CATALDO, September 2018.
from pylab import *
# Physical parameters
epsilon = 0.0103 # LJ potential Argon
sigma = 3.41 # LJ potential Argon
m = 39.948 # Argon Mass
dt=0.2 # in fs ( 1 fs = 10^-15 s)
# Lattice
nx=5
ny=5
a0 = 4.056
L = [ nx*a0, ny*a0 ]
# MD
nBlocks = 400
PBC = True # Periodic boundary conditions
r = array([ [a0*i,a0*j] for i in range(nx) for j in range(ny) ])
v = array([ [random(), random()] for i in range(nx) for j in range(ny) ]); v= (2*v-1)/20.0
a = array([ [0.0,0.0] for i in range(nx) for j in range(ny) ])
a_old = array([ [0.0,0.0] for i in range(nx) for j in range(ny) ])
'''
EXAMPLE 1:
'''
#nx=2; ny=1; dt= 2.0; a0 = 1.2*sigma; PBC= False; nBlocks= 400
#r = array([ [2, 2], [2+a0, 2] ])
#v = array([ [0.002,0.0], [-0.002,0.0] ] )
'''
EXAMPLE 2:
'''
dt = 1.0; nBlocks=400
v = array([ [random(), random()] for i in range(nx) for j in range(ny) ]); v= (2*v-1)/20.0
'''
EXAMPLE 3:
'''
#dt = 0.2
#v = array([ [random(), random()] for i in range(nx) for j in range(ny) ]); v= (2*v-1)/20.0
NATOMS= len(r)
def LennardJones(r): # r is the collection of all positions ri=(xi,yi)
U = 0.0
for i in range(NATOMS):
for j in range(i+1,NATOMS):
dr = r[i] -r[j]
if PBC:
if ( dr[0] >= 0.5*L[0] ): dr[0] -= L[0]
elif ( dr[0] < -0.5*L[0]): dr[0] += L[0]
if ( dr[1] >= 0.5*L[1] ): dr[1] -= L[1]
elif ( dr[1] < -0.5*L[1]): dr[1] += L[1]
rij = norm(dr)
U += 4.0*epsilon*( (sigma/rij)**12 - (sigma/rij)**6 )
return U
def KineticEnergy(v):
#v2 = [ dot(vi,vi) for vi in v ]
#sum_v2 = reduce((lambda x, y: x + y), v2)
#return 0.5*m*sum_v2*103.6427 # amu * (angstrom/fs)^2 = 103.6427 eV
v2 = 0.0
for vi in v:
v2 += norm(vi)**2
return 0.5*m*v2*103.6427
def Force(ri,rj):
dr = ri -rj
if PBC:
if ( dr[0] >= 0.5*L[0] ): dr[0] -= L[0]
elif ( dr[0] < -0.5*L[0]): dr[0] += L[0]
if ( dr[1] >= 0.5*L[1] ): dr[1] -= L[1]
elif ( dr[1] < -0.5*L[1]): dr[1] += L[1]
rij = norm(dr)
coeff = (sigma/rij)
force = -4*epsilon*( 12*coeff**11 - 6*coeff**5 ) * (-coeff/rij)*dr/norm(dr)
return force # in eV/angstrom
fig = figure(1)
ax = subplot(111)
xlim(-0.1, 1.1*L[0])
ylim(-0.1, 1.1*L[1])
#xlim(min(r[:,0]), max(r[:,0]),)
#ylim(2*(min(r[:,1]) -1), 2*(max(r[:,1])+1) )
p, = plot(r[:,0], r[:,1], 'o')
show(block=False)
"""
MOLECULAR DYNAMICS
"""
for iBlock in xrange(nBlocks):
''' PRINT PROPERTIES '''
print "t[fs]=",iBlock*dt, "Kin[eV]=", KineticEnergy(v), "Pot[eV]=", LennardJones(r)
for i in range(NATOMS):
a[i] = 0*a[i]
# Force over atom i
for j in range(NATOMS):
if i != j:
a[i] += 0.0096485333*Force(r[i],r[j])/m # <---- Inefficient, repited division and units conversion ( Force/mass = (eV/A)/(amu) = 0.0096485333 A/fs^2 )
'''
Euler-Cromer algorithm
'''
#####
v[i] = v[i] + dt*a[i]
r[i] = r[i] + dt*v[i]
####
'''
Velocity Verlet algorithm
'''
#####
#r[i] = r[i] + dt*v[i] + 0.5*dt*dt*a[i]
#v[i] = v[i] + dt*0.5*(a[i] + a_old[i])
#a_old[i] = a[i]
####
if PBC:
for q in range(2):
if r[i][q] >= L[q]: r[i][q] -= L[q]
elif r[i][q] <= 0.0: r[i][q] += L[q]
p.set_xdata(r[:,0])
p.set_ydata(r[:,1])
plt.pause(1e-30)
draw()
#savefig(str(iBlock)+'.png')